
People with dementia start to forget and often show changes in their abilities and personality. Over time the failure of short-term memory gradually turns into confusion about time and place, which may turn to depression or even aggressive behavior in later stages.
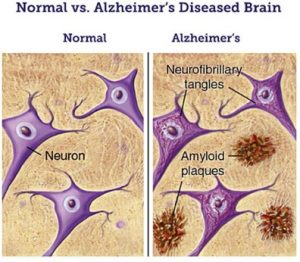
Normal vs Alzheimer’s disease brain (image from (6))
In principle, Alzheimer’s disease (AD) is an advanced stage of dementia that gets progressively worse over time. Alzheimer’s disease accounts for around 50-70% of all dementia cases in the EU (reported by European commission on Health and Food Safety). Currently, there is no cure for AD but there is a worldwide attempt to find better ways to delay its onset, treat the disease and hamper it from developing.
The major known risk factor for AD is advancing age (a non-modifiable risk and non-avoidable factor): the majority of people who are affected by Alzheimer are age 65 and older (1). There is strong evidence that cardiovascular risk factors such as hypertension, stroke, high cholesterol, and diabetes are also associated with AD. In a minority of cases, there is a familiar background with mutations in disease associated genes that can result in higher risk or earlier onset of the disease (1,2). While the origin of AD is not entirely known, there are two main neuropathological hallmarks known to cause Alzheimer’s disease. The first is amyloid beta peptides (Aβ), which aggregate outside the nerve cells forming amyloid plaques. Second, the present of tau proteins which undergo hyperphosphorylation and aggregate forming neurofibrillary tangles (NFT) inside the nerve cells (3).
Increasing evidence suggests that neuroinflammation has emerged as an important characteristic of AD pathology. Increased levels of pro-inflammatory mediators including complement activators and inhibitors, cytokines, chemokines, radical oxygen and inflammatory enzymes have been found in the brain of patients with Alzheimer’s disease, which illustrate the activation of a pathological inflammatory process (4,5).
Amyloid beta and plaque formation
Beta amyloid precursor protein (APP) is a crucial factor in AD pathogenesis. APP undergoes proteolysis by β-secretase and γ-secretase producing amyloid beta peptides (Aβ). Aβ is chemically “sticky” and gradually forms into plaques that are hallmarks of Alzheimer’s. The most common isoforms are Aβ40 and Aβ42. Aβ monomers can spontaneously assemble into Aβ oligomers (soluble) and fibrils (insoluble). It has been widely reported that soluble oligomers are the most toxic form of Aβ to neurons (5). Our body has a breakdown system to clear amyloid from the brain. However, in AD patients there is an imbalance between production and clearance, causing Aβ to accumulate. This imbalance is discussed as the earliest point of disease development, with multiple factors that all contribute to shift the balance from healthy aging to dementia. There are discussions that this accumulation can start early in healthy people, possibly already in the 30´s – but how and why this turns into disease decades later and why some people develop dementia in consequence while others don´t is still unanswered by research (5,7).
Toxic tau aggregation
Another common primary marker of AD is neurofibrillary tangles (NFTs). NFTs are aggregates of abnormally hyperphosphorylated tau protein, and in contrast to amyloid plaques, NFTs form inside the cell. Tau proteins are proteins that function as stabilizators of microtubules, a cellular transportation system. The loss of normal tau function, combined with a toxic gain of function – the aggregation – could compromise transport along the long nerve cell projections, the axons, and contribute to degeneration beginning with the important signaling synapses at the end of the axon. Furthermore, formation of large NFTs inside the cell can result in cell death and the number of neurofibrillary tangles is linked with the severity of AD. There is speculation that the formation of NFTs and amyloid aggregation are interlinked for example by toxic amyloid oligomers. The exact mechanism however is not finally resolved (3,8).
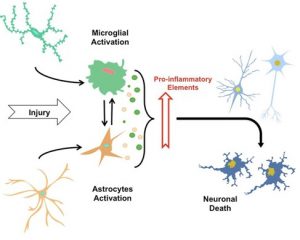
The neuroinflammatory process (modified from (9))
Neuroinflammation and AD
The term “neuroinflammation” is described as the inflammatory response in the central nervous system (CNS) after an injury that causes glial cells to accumulate. Alongside with their toxicity, Aβ deposition and tau protein cause synaptic function loss and activation of glial cells. Glial cells, i.e., microglia and astrocytes, but also neurons, are reacting and assisting to the neuroinflammatory changes in AD (4,5,9).
Microglia – the brain immune cells
The innate immune system is the first line of defense of the organism against different types of pathogens. Microglia cells are the key player of innate immunity in the CNS. At the same time, microglia also play a role in the maintenance and plasticity of neuronal circuits, adding synapses remodeling and protection. Once immunostimulated, these cells release a wide range of pro-inflammatory cytokines such as interleukin (IL)-1β, IL-6, tumor necrosis factor-α (TNF-α) and free radicals such as reactive oxygen species (ROS). This response is part of our defense against threats like invading microorganisms, but all of these molecules can also lead to neuronal damage and neuronal death. There are two known microglia phenotypes, M1 and M2, which are pro-inflammatory and neuroprotective, respectively. These phenotypes can interchange during the process of the disease (5,10,11).
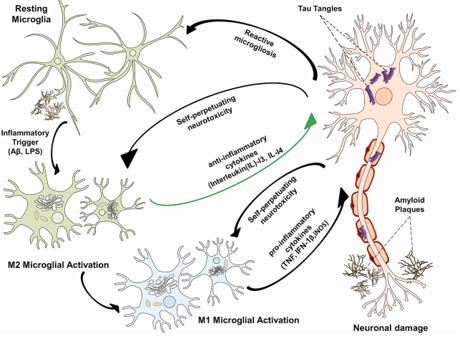
Relationship between amyloid plaques, neurofibrillary tangle and microglia activation (image from (11)).
Recent studies showed that an increased level of pro-inflammatory cytokines can play multiple roles in both neuroprotection and neurodegeneration. If neuroinflammation is not only a transient phenomenon – i.e. after injury or infection – but becomes a chronic process as in AD, this defense system does more damage than good especially to the vulnerable neurons. The questions: when, how, to which extend and by which mechanisms neuroinflammation contributes to the progress of AD and other dementias, are still ongoing research (5,10).
Astrocytes – star-shaped glia cell
Astrocytes are the most abundant glial cells in the brain (20% to 40% of all the glia cells). Astrocytes perform many functions: activation of neuronal synapses formation (synaptogenesis), blood-brain barrier maintenance, neurotransmission, metabolic regulation and play a key role in nervous system development. Therefore, loss of astroglial protective function contributes to neurodegenerative disease. Further, astrocytes are closely interacting with microglia and their interaction plays a main role in CNS inflammation (10,12). Thus, it is not surprising that pathological modifications of astrocytes have been related to several neurodegenerative disorders, including AD, Parkinson’s disease and multiple sclerosis. Alois Alzheimer actually already noticed the pathological modification of astrocytes in AD. He observed that glial cells are in close relation with damaged neurons, and that they are critical components of senile plaques (Alzheimer, 1910).
Over the past decade, increasing evidence suggests that neuroinflammation contributes to the process and pathogenesis of AD. The production and secretion of proinflammatory mediators may influence neurodegeneration at multiple levels. Hence a few proinflammatory cytokines can induce a neuropathic mechanism that leads to neuronal death, or can influence the neurodegenerative pathways. Finally, it is essential to pursue neuroinflammation as a pathway for a better understanding and cementing its connection to the pathological process in AD.
Author: Teodora Stella Wijasa
References:
1. Alzheimer’s & Dementia. 2016. Risk factors.
2. Risk factors for Alzheimer’s Disease. 2016, February 1.
3. Serrano-Pozo, A., Frosch, M.P., Masliah, E., Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb Perspect Med. 1(1):a006189 (2011). doi: 10.1101/cshperspect.a006189.
4. Heneka, M.T., O’banion, M.K., Terwel, D. & Kummer, M.P. Neuroinflamatory processes in Alzheimer‘s disease. J. Neural Transm. 117(8), 919-47 (2010). doi: 10.1007/s00702-010-0438-z. Epub 2010 Jul 15.
5. Heneka, M.T., Golenbock, D.T. & Latz, E. Innate immunity in Alzheimer’s disease. Nature Immunology. 16, 229-236 (2015). doi:10.1038/ni.3102.
6. Amyloid Plaques and Neurofibrillary tangles. 2000. Retrieved from brain focus foundation.
7. Tarasoff-Conway, J.M. et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 11(8), 457–470 (2015). doi:10.1038/nrneurol.2015.119.
8. Gendron, T.F. & Petrucelli, L. The role of tau in neurodegeneration. Mol Neurodegeneration. 4, 13 (2009). doi: 10.1186/1750-1326-4-13.
9. Morales, I., Guzman-Martinez, L., Cerda-Troncosco, C., Farias, G.A. & Maccioni, R.B. Neuroinflammation in the pathogenesis of alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front. Cell. Neurosci., 8, 112 (2014).
10. Heneka, M.T. & O’Banion, M.K. Inflammatory processes in Alzheimer’s disease. Jneuroim. 184, 69-91 (2007).
11. Fan, Z., Okello, A.A., Brooks, D.J. & Edison, P. Longitudinal influence of microglial activation and amyloid on neuronal function in Alzheimer’s disease. Brain. 138, 3685-3698 (2015). doi.org/10.1093/brain/awv288
12. Ricci, G., Volpi, L., Pasquali, L., Petrozzi, L. & Siciliano, G. Astrocyte-neuron interactions in neurological disorders. J Biol. Phys. 35, 317-336 (2009).doi:10.1007/s10867-009-9157-9.
(featured image from colourbox.com/ Kunertus)
Pingback: DZNE - Connecting the mental health of Rhineland people with basic research - ImmunosensationBlog