
In this blog article, I would like to talk about two-photon (2P) and three-photon (3P) in vivo imaging in the neuroscience field. However, prior to examining these new techniques, it is important to mention how the adventure in microscopy started. Microscopy plays a critical role in uncovering new dimensions in modern science.
The first compound microscope was invented by Zacharias and Hans Jansen in 1590, which was improved in 1609 by Galileo Galilei and named ‘’little eye’’. It is necessary here to clarify that the term ‘microscope’ was introduced for the first time by Giovanni Faber on 13 April 1625. Faber explained it as …novo occhiale di veder le cose minute, etlo chiamo microscopio “…I also mention its new eyeglass to look at minute things, and call it a microscope“1. Furthermore, in 1665 Robert Hooke published “Micrographia”, which was the first written document for the use of a microscope in natural science 2. In addition, Antony van Leeuwenhoek was the first person who observed bacteria and protozoa under the microscope in 16663(You can visit the cited web pages to see the microscopes built by Zacharias and Hans Jansen, Galileo Galilei, Robert Hooke and Antony van Leeuwenhoek 2,4–7).
These signs of progress in the microscope field allowed scientists for further investigations on biological samples and to make precise descriptions of cells, tissues, and organs. For example, Thomas Willis (known as the father of neuroscience) and Albrecht von Haller (known as the father of experimental physiology) investigated the structure of the nervous system and dynamics of neuro-muscular functions using microscope in the 17th century6.
From the 16th century to today, microscopy has improved remarkably, and we are now able to perform deep-tissue in vivo imaging with the help of high-power pulsed lasers. These instruments are particularly important for the field of neuroscience, as the brain is a very complex organ that harbors many different cell types including neurons, microglia, and astrocytes; in vivo imaging gives us a great opportunity to understand the interactions between these cells and to visualize their physiological and pathological changes in different conditions8.
Over the past two decades, most research in the field of neuroscience has emphasized the use of two-photon (2P) microscopy. In many directions, a 2P microscope bears resemblance to a confocal microscope. However, there are some important differences between 2P and confocal microscopes such as the excitation laser and the detection pathway. 2P microscopes can be converted from confocal microscopes (major modifications are needed) or can be custom-built, however, technical expertise is required to achieve it9,10.
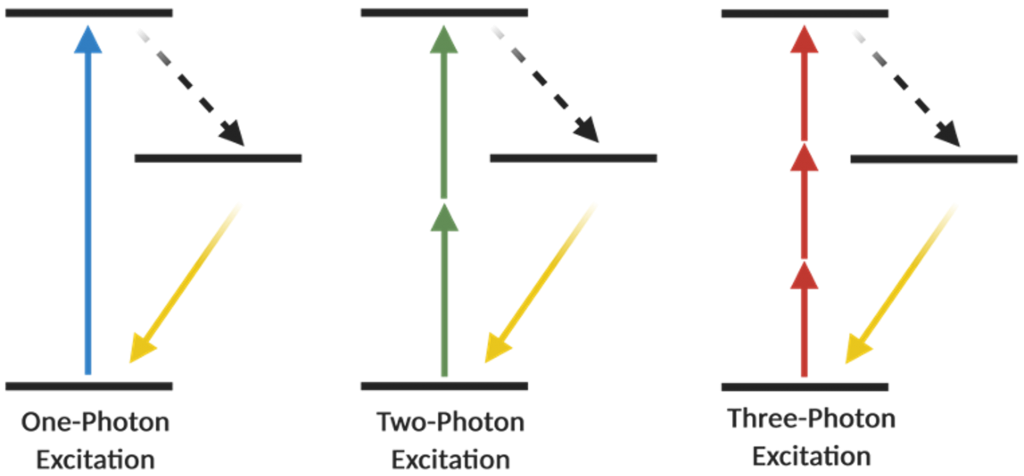
What do we need to know in order to have a better understanding for the backyard of 2P microscopy? As many of you know that a Jablonski energy diagram shows different energy levels, which take part in the absorption and emission of light by a fluorophore, and fluorescence microscopy mainly works on this rule. In the confocal imaging which is based on single-photon excitation, one photon (1P) has sufficient energy in order to excite an electron of a fluorophore to a higher energy state and after that excited electron relaxes by photon emission and go back into the ground-level energy state11. Maria Göppert-Mayer, the first scientist who described the concept of multi-photon excitation in 193112 (she received the Nobel Prize in Physics in 1963), proposed that if two or more photons arrive at a fluorophore simultaneously at the same time (within ~0.5 fs), and as long as their total energy is sufficient, then those photons can excite an electron to its higher energy levels. Subsequently, the electron relaxes, returns to its basal state and emits light, in other words, produces fluorescence signal11,13 (Figure 1). Around 60 years later, the first 2P microscope was invented by Winfried Denk and James Strickler in the lab of Watt W. Webb at Cornell University in 1990 and the first 2P images were acquired using Titan-Sapphire laser7. In order to meet 2P excitation criteria, lasers play an important role and for 2P microscopy, mode-locked Titan-Sapphire lasers have ideal properties to pulse light with a repetition rate of 70–100 MHz and a pulse width of 70–100 fs (10−15 s)11,14.
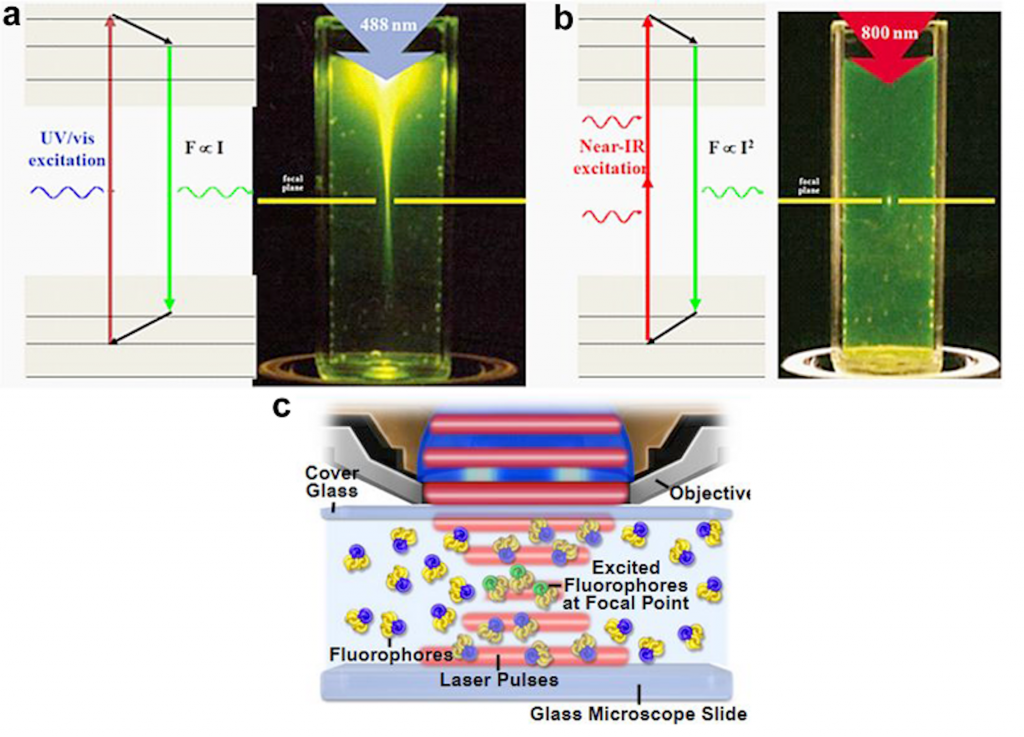
There are a number of important advantages in 2P in vivo microscopy when it is compared with confocal microscopy. One of them is light scattering in tissue samples. In the 2P microscopy, the excitation wavelength is between 700 to 1000 nm (near-infrared wavelength range), which gives the researchers an opportunity to excite the most beneficial fluorophores14. Due to the fact that cells consist of a heterogeneous mixture of various molecules, light scattering is more prone in biological tissues9. As stated above, 2P excitation happens in near-infrared wavelength range, and this provides a better tissue penetration, a reduced scattering, and an increased resolution16. Using 2P microscopy high-resolution deep tissue imaging is possible below an organ surface17, while confocal imaging does not give this possibility. One major issue in confocal microscopy is phototoxicity too. In a confocal microscope, there is a pinhole where the emitted light passes through, in order to eliminate out of focus fluorescent light. On the other hand in 2P microscopy the excitation only occurs at the focal point and having a pinhole is not needed. By this way, less amount of tissue is affected by higher energy levels, which decreases possible photobleaching and photodamage in the imaging area11. Using 2P microscopy both awake and anesthetized in vivo imaging sessions are possible in living animals. Today, resolution improvements for 2P microscopy are still in progress. Recently, 2P microscopy has been combined with super-resolution stimulated emission depletion (STED) microscopy and this combination increased the spatial resolution for in vivo dendritic spine imaging in living mice 18.
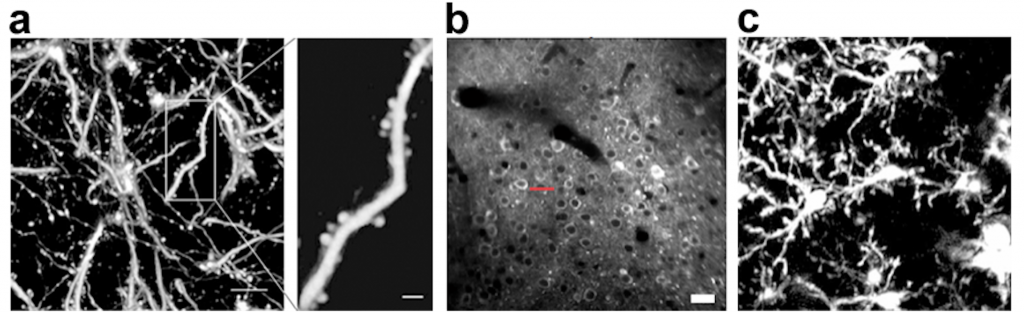
In vivo 2P microscopy is a powerful technique especially in the field of neuroscience. Nowadays, we are able to measure neuronal network activity by calcium imaging, examine synaptic plasticity by dendritic spine imaging, investigate inflammatory responses by microglial imaging, and visualize neurotransmitters using various fluorescent sensors in living brains (Figure 2), etc.
2P microscopy solved many issues that confocal microscopy could not achieve, nevertheless high-resolution tissue imaging in much deeper levels without invasive applications such as removing part of the cortex or inserting optical lenses (GRIN lenses, etc)22,23, was still needed. These technical limitations can be overcome by three-photon (3P) microscopy. Unlike 2P microscopy, in this technique, three photons are needed for the excitation (Figure 3). One of the important scientists in the development of 3P microscopy is Chris Xu. When Chris Xu was a PhD student in Watt Webb’s lab, they proved 3P fluorescence imaging in 1995.
3P excitation brings significant advantages in contrast to 2P excitation. In deeper tissue levels light scattering increases. Due to the fact that 3P excitation uses a more broad range of wavelength (up to 1700 nm), this strategy provides increased tissue penetration and decreased light scattering. Furthermore, 3P microscopy enables us to take high-contrast images up to ~1300 um deep in intact mouse brain without any invasive application. For instance CA1 region, which is ~956 below the surface of the brain, can be imaged comfortably at high resolution by cortical cranial window24,25. In addition, 3P microscopy provides third-harmonic generation signal (THG). Here, the energy of three photons is combined during excitation and afterwards converted into single emitted photon26, enabling label-free imaging for blood vessels and myelinated axons 25,27. It is beneficial because extra structural information can be obtained without any additional staining. 3P microscopy was used only in anesthetized animals in intact mouse brain until recently as it was combined with awake imaging by Mriganka Sur group27. Moreover, Jason Kerr`s lab advanced it one step further by designing a head mounted 3P microscope in freely moving rats 28.
Every piece of new development increases our understanding. For example, if in vivo imaging technique had not been invented, we would not be able to observe the brain in living animals. If a picture is worth a thousand words, one video recorded from a living brain is priceless. Maybe in the future, we will be able to image the whole brain without any depth limitation in live animals. Therefore, as Queen (the British rock band) said ‘’ The Show Must Go On’’.
Author: Dilek Mercan
Acknowledgements
This work was supported by SFB 1089‚ Synaptic Micronetworks in Health and Disease. Thanks to Dr. Heela Sarlus for proofreading this blog article.
D. Mercan was supported with a ImmunoSensation Travel-and Workshop Grant
References
1. Bardell, D. The invention of the microscope. 75, 8 (2004).
2. National Museums Scotland. Early Microscopes. https://www.nms.ac.uk/explore-our-collections/stories/science-and-technology/microscopes/microscopes-chapters/early-microscopes/.
3. The Editors of Encyclopaedia Britannica. Antonie van Leeuwenhoek. https://www.britannica.com/biography/Antonie-van-Leeuwenhoek (2019).
4. NIH. Hooke’s Books. https://www.nlm.nih.gov/exhibition/hooke/micrographia.html.
5. History of Science Museum. Leeuwenhoek Replica Microscope. http://www.mhs.ox.ac.uk/collections/imu-search-page/record-details/?TitInventoryNo=41858&querytype=field&thumbnails=on.
6. Compound microscope, Galilean. https://catalogue.museogalileo.it/object/CompoundMicroscopeGalilean.html.
7. Who Invented the Microscope? https://www.livescience.com/39649-who-invented-the-microscope.html.
8. Mercan, D. & Heneka, M. T. Norepinephrine as a modulator of microglial dynamics. Nat Neurosci22, 1745–1746 (2019).
9. Helmchen, F. & Denk, W. Deep tissue two-photon microscopy. Nat Methods 2, 932–940 (2005).
10. Denk, W., Strickler, J. H. & Webb, W. W. Two-Photon Laser Scanning Fluorescence Microscopy. Science, New Series 248, 73–76 (1990).
11. Dimitrios Davalos & Martin Fuhrmann. Chapter 4: Lessons from In Vivo Imaging. in Microglia in health and disease (eds. Tremblay, M.-È. & Sierra, A.) (Springer, 2014).
12. Göppert-Mayer, M. Über Elementarakte mit zwei Quantensprüngen. Ann. Phys. 401, 273–294 (1931).
13. David W. Piston, Thomas J. Fellers, & Michael W. Davidson. Multiphoton Microscopy: Fundamentals and Applications in Multiphoton Excitation Microscopy. MolecularExpressions.com at Florida State Universityhttps://micro.magnet.fsu.edu.
14. Svoboda, K. & Yasuda, R. Principles of Two-Photon Excitation Microscopy and Its Applications to Neuroscience. Neuron 50, 823–839 (2006).
15. Zipfel, W. R., Williams, R. M. & Webb, W. W. Nonlinear magic: multiphoton microscopy in the biosciences. Nat Biotechnol 21, 1369–1377 (2003).
16. Oheim, M., Beaurepaire, E., Chaigneau, E., Mertz, J. & Charpak, S. Two-photon microscopy in brain tissue: parameters influencing the imaging depth. Journal of Neuroscience Methods 111, 29–37 (2001).
17. Theer, P., Hasan, M. T. & Denk, W. Two-photon imaging to a depth of 1000 µm in living brains by use of a Ti:Al2O3 regenerative amplifier. 3.
18. Pfeiffer, T. et al. Chronic 2P-STED imaging reveals high turnover of dendritic spines in the hippocampus in vivo. eLife 7, e34700 (2018).
19. Fuhrmann, M., Mitteregger, G., Kretzschmar, H. & Herms, J. Dendritic Pathology in Prion Disease Starts at the Synaptic Spine. Journal of Neuroscience 27, 6224–6233 (2007).
20. Akerboom, J. et al. Optimization of a GCaMP Calcium Indicator for Neural Activity Imaging. Journal of Neuroscience 32, 13819–13840 (2012).
21. Davalos, D. et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8, 752–758 (2005).
22. Dombeck, D. A., Harvey, C. D., Tian, L., Looger, L. L. & Tank, D. W. Functional imaging of hippocampal place cells at cellular resolution during virtual navigation. Nat Neurosci 13, 1433–1440 (2010).
23. Ziv, Y. et al. Long-term dynamics of CA1 hippocampal place codes. Nat Neurosci 16, 264–266 (2013).
24. Horton, N. G. et al. In vivo three-photon microscopy of subcortical structures within an intact mouse brain. Nature Photon 7, 205–209 (2013).
25. Ouzounov, D. G. et al. In vivo three-photon imaging of activity of GCaMP6-labeled neurons deep in intact mouse brain. Nat Methods 14, 388–390 (2017).
26. Weigelin, B., Bakker, G.-J. & Friedl, P. Third harmonic generation microscopy of cells and tissue organization. J Cell Sci 129, 245–255 (2016).
27. Yildirim, M., Sugihara, H., So, P. T. C. & Sur, M. Functional imaging of visual cortical layers and subplate in awake mice with optimized three-photon microscopy. Nat Commun 10, 177 (2019).
28. Klioutchnikov, A. et al. Three-photon head-mounted microscope for imaging deep cortical layers in freely moving rats. Nature Methods 17, 509–513 (2020).